



Récemment, trois médicaments de thérapie génique ont été approuvés pour la commercialisation, à savoir : (1) Le 21 juillet 2022, PTC Therapeutics, Inc. (NASDAQ : PTCT) a annoncé que sa thérapie génique AAV Upstaza™ a été approuvée par la Commission européenne. Il s'agit de la première thérapie génique commercialisée directement injectée dans le cerveau (voir articles précédents : Another Milestone in Gene Therapy |(2) Le 17 août 2022, la Food and Drug Administration (FDA) des États-Unis a approuvé la thérapie génique Zynteglo (betibeglogene autotemcel, beti-cel) de Bluebird Bio pour le traitement de la bêta-thalassémie.L'approbation de la thérapie aux États-Unis est sans aucun doute une "aide dans la neige" pour Bluebird Bio, qui est en crise financière.(3) Le 24 août 2022, BioMarin Pharmaceutical (BioMarin) a annoncé que la Commission européenne avait autorisé la mise sur le marché conditionnelle de ROCTAVIAN™ (valoctocogene roxaparvovec), une thérapie génique de l'hémophilie A, pour le traitement des patients sans antécédents d'inhibiteurs du facteur FVIII et d'anticorps AAV5 négatifs chez les patients adultes atteints d'hémophilie A sévère (voir article précédent : La thérapie génique de l'hémophilie A de Heavy! BioMarin approuvée pour la commercialisation).Jusqu'à présent, 41 médicaments de thérapie génique ont été approuvés pour la commercialisation dans le monde.
Le gène est l'unité génétique de base qui contrôle les traits.À l'exception des gènes de certains virus, qui sont composés d'ARN, les gènes de la plupart des organismes sont composés d'ADN.La plupart des maladies de l'organisme sont causées par l'interaction entre les gènes et l'environnement, et de nombreuses maladies peuvent être guéries ou soulagées essentiellement grâce à la thérapie génique.La thérapie génique est considérée comme une révolution dans le domaine de la médecine et de la pharmacie.Les médicaments de thérapie génique au sens large comprennent les médicaments à base d'ADN modifié par l'ADN (tels que les médicaments de thérapie génique in vivo basés sur un vecteur viral, les médicaments de thérapie génique in vitro, les médicaments à base de plasmide nu, etc.) et les médicaments à base d'ARN (tels que les médicaments à base d'oligonucléotides antisens, les médicaments à ARNsi et la thérapie génique à ARNm, etc.);Les médicaments de thérapie génique étroitement définis comprennent principalement les médicaments à base d'ADN plasmidique, les médicaments de thérapie génique basés sur des vecteurs viraux, les médicaments de thérapie génique basés sur des vecteurs bactériens, les systèmes d'édition de gènes et les médicaments de thérapie cellulaire génétiquement modifiés in vitro.Après des années de développement tortueux, les médicaments de thérapie génique ont obtenu des résultats cliniques inspirants.(hors vaccins ADN et vaccins ARNm), 41 médicaments de thérapie génique ont été approuvés pour commercialisation dans le monde.Avec le lancement de produits et le développement rapide de la technologie de thérapie génique, la thérapie génique est sur le point d'inaugurer une période de développement rapide.
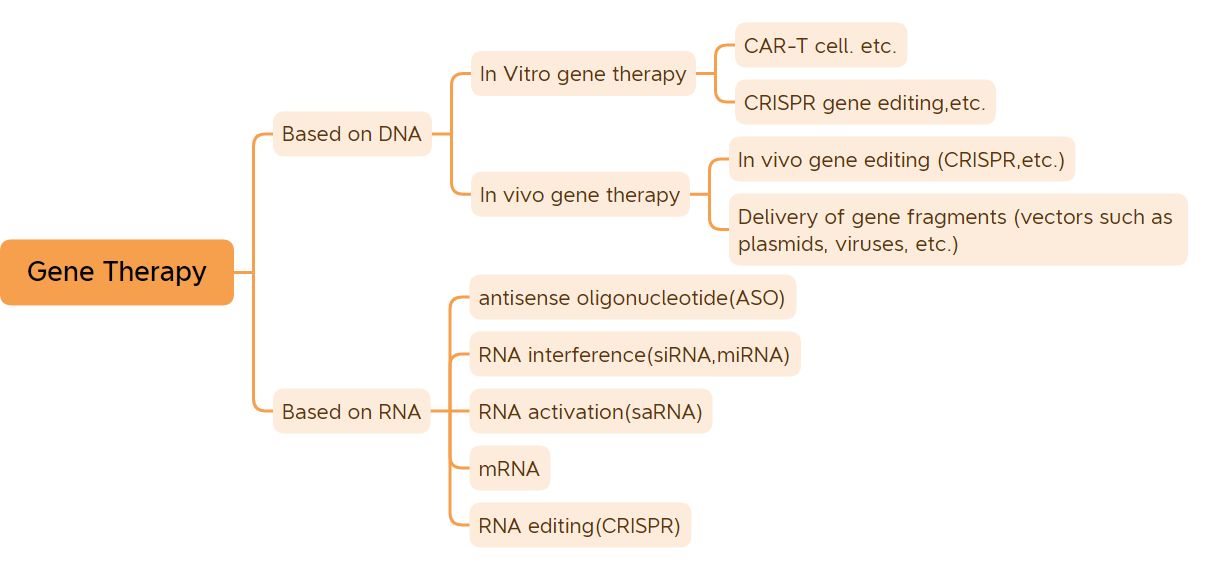
Classification de la thérapie génique (Source de l'image : Biological Jingwei)
Cet article répertorie 41 thérapies géniques dont la commercialisation a été approuvée (hors vaccins à ADN et vaccins à ARNm).
1. Thérapie génique in vitro
(1) Strimvélis
Entreprise : Développé par GlaxoSmithKline (GSK).
Délai de mise sur le marché : Approuvé par l'Union européenne en mai 2016.
Indications : Pour le traitement du déficit immunitaire combiné sévère (SCID).
Remarques : Le processus général de cette thérapie consiste à obtenir d'abord les propres cellules souches hématopoïétiques du patient, à les développer et à les cultiver in vitro, puis à utiliser un rétrovirus pour introduire une copie du gène fonctionnel ADA (adénosine désaminase) dans leurs cellules souches hématopoïétiques, et enfin à transférer les cellules souches hématopoïétiques modifiées.Les cellules souches hématopoïétiques sont réinjectées dans le corps.Les résultats cliniques ont montré que le taux de survie à 3 ans des patients ADA-SCID traités avec Strimvelis était de 100 %.
(2) Zalmoxis
Entreprise : Produit par MolMed, Italie.
Délai de mise sur le marché : A obtenu l'autorisation de mise sur le marché conditionnelle de l'UE en 2016.
Indications : Il est utilisé pour le traitement adjuvant du système immunitaire des patients après une greffe de cellules souches hématopoïétiques.
Remarques : Zalmoxis est une immunothérapie par gène suicide des cellules T allogéniques modifiées par un vecteur rétroviral.Les gènes suicides 1NGFR et HSV-TK Mut2 permettent aux gens d'utiliser le ganciclovir à tout moment pour tuer les lymphocytes T qui provoquent des réponses immunitaires indésirables, prévenir une nouvelle détérioration de la GVHD qui peut survenir et restaurer la fonction immunitaire chez les patients atteints de GCSH haploidentique après une intervention chirurgicale Escort.
(3) Invossa-K
Société : Développé par la société TissueGene (KolonTissueGene).
Délai de mise sur le marché : Approuvé pour cotation en Corée du Sud en juillet 2017.
Indications : Pour le traitement de l'arthrite dégénérative du genou.
Remarques : Invossa-K est une thérapie génique cellulaire allogénique impliquant des chondrocytes humains.Les cellules allogéniques sont génétiquement modifiées in vitro, et les cellules modifiées peuvent exprimer et sécréter le facteur de croissance transformant β1 (TGF-β1) après injection intra-articulaire.β1), améliorant ainsi les symptômes de l'arthrose.Les résultats cliniques montrent qu'Invossa-K peut améliorer de manière significative l'arthrite du genou.La licence a été révoquée par le régulateur sud-coréen des médicaments en 2019 parce que le fabricant a mal étiqueté les ingrédients utilisés.
(4) Zyntéglo
Société : Développé par la société américaine bluebird bio (bluebird bio).
Délai de mise sur le marché : Approuvé par l'Union européenne en 2019 et approuvé par la FDA en août 2022.
Indications : Pour le traitement de la β-thalassémie dépendante des transfusions.
Remarques : Zynteglo est une thérapie génique lentivirale in vitro, qui utilise un vecteur lentiviral pour introduire une copie fonctionnelle du gène normal de la β-globine (gène βA-T87Q-globine) dans des cellules souches hématopoïétiques prélevées sur des patients., puis réinsuffler ces cellules souches hématopoïétiques autologues génétiquement modifiées au patient.Une fois que le patient a un gène βA-T87Q-globine normal, il peut produire une protéine HbAT87Q normale, qui peut réduire ou éliminer efficacement le besoin de transfusions sanguines.Il s'agit d'un traitement unique conçu pour remplacer les transfusions sanguines à vie et les médicaments à vie pour les patients de 12 ans et plus.
(5) Skysona
Société : Développé par la société américaine bluebird bio (bluebird bio).
Délai de mise sur le marché : Approuvé par l'UE pour commercialisation en juillet 2021.
Indications : Pour le traitement de l'adrénoleucodystrophie cérébrale précoce (CALD).
Remarques : La thérapie génique Skysona est la seule thérapie génique ponctuelle approuvée pour le traitement de l'adrénoleucodystrophie cérébrale précoce (CALD).Skysona (elivaldogene autotemcel, Lenti-D) est une thérapie génique in vitro lentivirale de cellules souches hématopoïétiques Lenti-D.Le processus général de la thérapie est le suivant : des cellules souches hématopoïétiques autologues sont prélevées sur le patient, modifiées in vitro par un lentivirus portant le gène humain ABCD1, puis réinjectées au patient.Pour le traitement des patients de moins de 18 ans présentant une mutation du gène ABCD1 et CALD.
(6) Kymriah
Entreprise : Développé par Novartis.
Délai de mise sur le marché : Approuvé par la FDA en août 2017.
Indications : Traitement de la leucémie aiguë lymphoblastique (LAL) à précurseurs B et du DLBCL récidivant et réfractaire.
Remarques : Kymriah est un médicament de thérapie génique in vitro lentiviral, la première thérapie CAR-T approuvée au monde, ciblant CD19 et utilisant le facteur de co-stimulation 4-1BB.Le prix est de 475 000 $ aux États-Unis et de 313 000 $ au Japon.
(7) Yescarta
Entreprise : Développé par Kite Pharma, une filiale de Gilead.
Délai de mise sur le marché : Approuvé par la FDA en octobre 2017.
Indications : Pour le traitement du lymphome à grandes cellules B récidivant ou réfractaire.
Remarques : Yescarta est une thérapie génique rétrovirale in vitro.Il s'agit de la deuxième thérapie CAR-T approuvée dans le monde.Il cible CD19 et utilise le facteur costimulateur de CD28.Le prix aux États-Unis est de 373 000 $.
(8) Técartus
Entreprise : Développé par Gilead (GILD).
Délai de mise sur le marché : Approuvé par la FDA en juillet 2020.
Indications : Pour le lymphome à cellules du manteau récidivant ou réfractaire.
Remarques : Tecartus est une thérapie cellulaire CAR-T autologue ciblant CD19, et est la troisième thérapie CAR-T approuvée pour la commercialisation dans le monde.
(9) Breyanzi
Entreprise : Développé par Bristol-Myers Squibb (BMS).
Délai de mise sur le marché : Approuvé par la FDA en février 2021.
Indications : Lymphome à grandes cellules B (LBCL) récidivant ou réfractaire (R/R).
Remarques : Breyanzi est une thérapie génique in vitro à base de lentivirus, et la quatrième thérapie CAR-T dont la commercialisation est approuvée dans le monde, ciblant le CD19.L'approbation de Breyanzi est une étape importante pour Bristol-Myers Squibb dans le domaine de l'immunothérapie cellulaire, que Bristol-Myers a acquise lors de l'acquisition de Celgene pour 74 milliards de dollars en 2019.
(10) Abecma
Entreprise : Co-développé par Bristol-Myers Squibb (BMS) et bluebird bio.
Délai de mise sur le marché : Approuvé par la FDA en mars 2021.
Indications : Myélome multiple récidivant ou réfractaire.
Remarques : Abecma est une thérapie génique in vitro à base de lentivirus, la première thérapie cellulaire CAR-T au monde ciblant le BCMA et la cinquième thérapie CAR-T approuvée par la FDA.Le principe du médicament est d'exprimer le récepteur chimérique BCMA sur les lymphocytes T autologues du patient par modification génétique médiée par des lentivirus in vitro.Avant la perfusion du médicament génique cellulaire, le patient a reçu deux composés de cyclophosphamide et de fludarabine pour le prétraitement.Traitement pour éliminer les lymphocytes T non modifiés du patient, puis réinjecter les lymphocytes T modifiés dans le corps du patient pour rechercher et tuer les cellules cancéreuses exprimant le BCMA.
(11) Libmeldy
Entreprise : Développé par Orchard Therapeutics.
Délai de mise sur le marché : Approuvé par l'Union européenne pour cotation en décembre 2020.
Indications : Pour le traitement de la leucodystrophie métachromatique (MLD).
Remarques : Libmeldy est une thérapie génique basée sur la modification génique lentivirale in vitro de cellules CD34+ autologues.Les données cliniques montrent qu'une seule perfusion intraveineuse de Libmeldy est efficace pour modifier l'évolution de la MLD précoce et des troubles moteurs et cognitifs sévères chez les patients non traités du même âge.
(12) Bénoda
Entreprise : Développé par WuXi Junuo.
Délai de mise sur le marché : Officiellement approuvé par la NMPA en septembre 2021.
Indications : Traitement du lymphome à grandes cellules B récidivant ou réfractaire (LBCL r/r) chez les patients adultes après un traitement de deuxième ligne ou plus systémique.
Remarques : Benoda est une thérapie génique anti-CD19 CAR-T, et c'est également le produit phare de WuXi Junuo.Il s'agit du deuxième produit CAR-T approuvé en Chine, à l'exception du lymphome à grandes cellules B récidivant/réfractaire.En outre, WuXi Junuo prévoit également de développer l'injection de Ruiki Orenza pour le traitement de diverses autres indications, notamment le lymphome folliculaire (LF), le lymphome à cellules du manteau (MCL), la leucémie lymphoïde chronique (LLC), le lymphome diffus à grandes cellules B de deuxième intention (DLBCL) et la leucémie aiguë lymphoblastique (LAL).
(13) CARVYKTI
Entreprise : premier produit approuvé par Legend Bio.
Délai de mise sur le marché : Approuvé par la FDA en février 2022.
Indications : Traitement du myélome multiple récidivant ou réfractaire (R/R MM).
Remarques : CARVYKTI (ciltacabtagene autoleucel, appelé Cilta-cel) est une thérapie génique immunitaire cellulaire CAR-T avec deux anticorps à domaine unique ciblant l'antigène de maturation des cellules B (BCMA).Les données montrent que CARVYKTI a démontré un taux de réponse global allant jusqu'à 98 % chez les patients atteints de myélome multiple récidivant ou réfractaire qui avaient reçu au moins quatre traitements antérieurs, y compris des inhibiteurs du protéasome, des immunomodulateurs et des anticorps monoclonaux anti-CD38.
2. Thérapie génique in vivo basée sur des vecteurs viraux
(1) Gendicine/né de nouveau
Entreprise : Développé par Shenzhen Saibainuo Company.
Délai de mise sur le marché : Approuvé pour la cotation en Chine en 2003.
Indications : Pour le traitement du carcinome épidermoïde de la tête et du cou.
Remarques : injection d'adénovirus humain recombinant p53 Gendicine/Jinshengsheng est un médicament de thérapie génique à vecteur d'adénovirus avec des droits de propriété intellectuelle indépendants appartenant à la société Shenzhen Saibainuo.Le médicament est composé du gène suppresseur de tumeur humain normal p53 et de l'adénovirus humain de type 5 recombinant artificiellement modifié déficient en réplication est composé d'adénovirus humain de type 5. Le premier est la structure principale du médicament pour exercer un effet antitumoral, et le dernier agit principalement comme un transporteur.Le vecteur adénoviral porte le gène thérapeutique p53 dans la cellule cible et exprime le gène suppresseur de tumeur p53 dans la cellule cible.Le produit peut réguler à la hausse divers gènes anticancéreux et réguler à la baisse les activités de divers oncogènes, améliorant ainsi l'effet antitumoral du corps et atteignant l'objectif de tuer les tumeurs.
(2) Rigvir
Société : Développé par la société lettone Latima.
Time to market : Approuvé en Lettonie en 2004.
Indications : Pour le traitement du mélanome.
Remarques : Rigvir est une thérapie génique basée sur un vecteur d'entérovirus ECHO-7 modifié génétiquement, qui a été utilisé en Lettonie, en Estonie, en Pologne, en Arménie, en Biélorussie et ailleurs, et est également enregistré auprès de l'EMA de l'Union européenne..Des cas cliniques au cours des dix dernières années ont prouvé que le virus oncolytique Rigvir est sûr et efficace et peut améliorer de 4 à 6 fois le taux de survie des patients atteints de mélanome.En outre, la thérapie convient également à une variété d'autres cancers, notamment le cancer colorectal, le cancer du pancréas et le cancer de la vessie.cancer, cancer du rein, cancer de la prostate, cancer du poumon, cancer de l'utérus, lymphosarcome, etc.
(3) Oncorine/Ankerui
Entreprise : Développé par Shanghai Sunway Biotechnology Co., Ltd.
Délai de mise sur le marché : Approuvé pour la cotation en Chine en 2005.
Indications : Traitement des tumeurs de la tête et du cou, cancer du foie, cancer du pancréas, cancer du col de l'utérus et autres cancers.
Remarques : Oncorine est un produit de thérapie génique virale oncolytique utilisant l'adénovirus comme vecteur.L'adénovirus oncolytique obtenu peut se répliquer spécifiquement dans des tumeurs dépourvues ou anormales du gène p53, provoquant la lyse des cellules tumorales, tuant ainsi les cellules tumorales.sans endommager les cellules normales.Les résultats cliniques montrent qu'Anke Rui a une bonne sécurité et efficacité pour une variété de tumeurs malignes.
(4) Glybera
Entreprise : Développé par uniQure.
Time to market : Approuvé en Europe en 2012.
Indications : Traitement du déficit en lipoprotéine lipase (LPLD) avec épisodes sévères ou récurrents de pancréatite malgré un régime alimentaire strictement limité en matières grasses.
Remarques : Glybera (alipogene tiparvovec) est un médicament de thérapie génique basé sur l'AAV comme vecteur.Cette thérapie utilise l'AAV comme vecteur pour transférer le gène thérapeutique LPL dans les cellules musculaires, afin que les cellules correspondantes puissent produire une certaine quantité de lipoprotéine lipase. Elle joue un rôle dans le soulagement de la maladie, et cette thérapie est efficace pendant longtemps après une administration (l'effet peut durer de nombreuses années).Le médicament a été retiré de la liste en 2017 et les raisons de sa radiation peuvent être liées à deux facteurs : des prix trop élevés et une demande limitée du marché.Le coût moyen d'un seul traitement du médicament s'élève à 1 million de dollars américains, et un seul patient l'a acheté et utilisé jusqu'à présent.Bien que la compagnie d'assurance médicale l'ait remboursé pour 900 000 dollars américains, c'est aussi une lourde charge pour la compagnie d'assurance.De plus, l'indication du médicament est trop rare, avec un taux d'incidence d'environ 1 sur 1 million et un taux élevé d'erreurs de diagnostic.
(5) Imlygique
Entreprise : Développé par Amgen.
Délai de mise sur le marché : En 2015, il a été approuvé pour être coté aux États-Unis et dans l'Union européenne.
Indications : Traitement des lésions de mélanome qui ne peuvent pas être complètement éliminées par chirurgie.
Remarques : Imlygic est un virus oncolytique atténué du virus de l'herpès simplex de type 1 (HSV-1) génétiquement modifié (suppression de ses fragments de gènes ICP34.5 et ICP47 et insertion du gène GM-CSF du facteur stimulant les colonies de granulocytes-macrophages humains), la première thérapie génique par virus oncolytique approuvée par la FDA.Le mode d'administration est l'injection intralésionnelle.L'injection directe dans les lésions de mélanome peut provoquer la rupture des cellules tumorales et libérer des antigènes dérivés de la tumeur et du GM-CSF pour favoriser les réponses immunitaires anti-tumorales.
(6) Luxturna
Entreprise : Développé par Spark Therapeutics, une filiale de Roche.
Time to market : Approuvé par la FDA en 2017, puis approuvé pour la commercialisation en Europe en 2018.
Indications : Pour le traitement des enfants et des adultes présentant une perte de vision due à des mutations de la double copie du gène RPE65 mais avec un nombre suffisant de cellules rétiniennes viables.
Remarques : Luxturna est une thérapie génique basée sur l'AAV qui est administrée par injection sous-rétinienne.La thérapie génique utilise AAV2 comme porteur pour introduire une copie fonctionnelle du gène RPE65 normal dans les cellules rétiniennes du patient, de sorte que les cellules correspondantes expriment la protéine RPE65 normale pour compenser le défaut de la protéine RPE65 du patient, améliorant ainsi la vision du patient.
(7) Zolgensme
Entreprise : Développé par AveXis, une filiale de Novartis.
Délai de mise sur le marché : Approuvé par la FDA en mai 2019.
Indications : Traitement des patients atteints d'amyotrophie spinale (amyotrophie spinale, SMA) de moins de 2 ans.
Remarques : Zolgensma est une thérapie génique basée sur le vecteur AAV.Ce médicament est le seul plan de traitement unique pour l'amyotrophie spinale dont la commercialisation a été approuvée dans le monde.page, est un progrès d'étape.Cette thérapie génique utilise le vecteur scAAV9 pour introduire le gène SMN1 normal chez les patients par perfusion intraveineuse, produisant la protéine SMN1 normale, améliorant ainsi la fonction des cellules affectées telles que les motoneurones.En revanche, les médicaments SMA Spinraza et Evrysdi nécessitent des doses répétées sur une longue période de temps, avec Spinraza administré sous forme d'injection spinale tous les quatre mois, et Evrysdi, un médicament oral quotidien.
(8) Delytacte
Entreprise : Développé par Daiichi Sankyo Company Limited (TYO : 4568).
Délai de mise sur le marché : approbation conditionnelle du ministère japonais de la Santé, du Travail et des Affaires sociales (MHLW) en juin 2021.
Indications : Pour le traitement du gliome malin.
Remarques : Delytact est le quatrième produit de thérapie génique par virus oncolytique approuvé dans le monde et le premier produit viral oncolytique approuvé pour le traitement du gliome malin.Delytact est un virus oncolytique génétiquement modifié du virus de l'herpès simplex de type 1 (HSV-1) développé par le Dr Todo et ses collègues.Delytact introduit une mutation par délétion supplémentaire dans le génome G207 du HSV-1 de deuxième génération, améliorant sa réplication sélective dans les cellules cancéreuses et l'induction de réponses immunitaires anti-tumorales, tout en maintenant un profil de sécurité élevé.Delytact est le premier HSV-1 oncolytique de troisième génération actuellement en évaluation clinique.L'approbation de Delytact au Japon était basée sur un essai clinique de phase 2 à un seul bras.Chez les patients atteints de glioblastome récurrent, Delytact a atteint le critère principal de survie à un an, et les résultats ont montré que Delytact était plus performant que G207.Forte réplication et activité antitumorale plus élevée.Ceci est efficace dans les modèles de tumeurs solides, notamment les tumeurs du sein, de la prostate, du schwannome, du nasopharynx, des cellules hépatocellulaires, colorectales, des gaines nerveuses périphériques malignes et le cancer de la thyroïde.
(9) Upstaza
Société : Développé par PTC Therapeutics, Inc. (NASDAQ : PTCT).
Délai de mise sur le marché : Approuvé par l'UE en juillet 2022.
Indication : Pour le déficit en décarboxylase des acides aminés L aromatiques (AADC), approuvé pour le traitement des patients âgés de 18 mois et plus.
Remarques : Upstaza™ (eladocagene exuparvovec) est une thérapie génique in vivo utilisant le virus adéno-associé de type 2 (AAV2) comme vecteur.Le patient est malade en raison d'une mutation du gène codant pour l'enzyme AADC.AAV2 porte un gène sain codant pour l'enzyme AADC.L'effet thérapeutique est obtenu sous la forme d'une compensation génétique.En théorie, une dose unique est efficace pendant longtemps.Il s'agit de la première thérapie génique commercialisée directement injectée dans le cerveau.L'autorisation de mise sur le marché s'applique aux 27 États membres de l'UE, ainsi qu'à l'Islande, la Norvège et le Liechtenstein.
(9) Roctave
Société : Développé par BioMarin Pharmaceutical (BioMarin).
Délai de mise sur le marché : Approuvé par l'UE en août 2022.
Indications : Pour le traitement des patients adultes atteints d'hémophilie A sévère sans antécédent d'inhibition du facteur FVIII et anticorps AAV5 négatifs.
Remarques : Roctavian (valoctocogene roxaparvovec) utilise AAV5 comme vecteur et utilise le promoteur spécifique du foie humain HLP pour piloter l'expression du facteur de coagulation humain huit (FVIII) avec le domaine B supprimé.La décision de la Commission européenne d'autoriser la commercialisation du valoctocogène roxaparvovec est basée sur les données globales du programme de développement clinique du médicament.Parmi eux, l'essai clinique de phase III GENEr8-1 a montré que par rapport aux données de l'année précédant l'inscription, après une seule perfusion de valoctocogène roxaparvovec, les sujets présentaient un taux de saignement annuel (ABR) significativement plus faible, une utilisation moins fréquente de préparations de protéines recombinantes de facteur VIII (F8), ou une augmentation significative de l'activité F8 dans le sang de l'organisme.Après 4 semaines de traitement, l'utilisation annuelle de F8 des sujets et l'ABR nécessitant un traitement ont été réduits de 99 % et 84 %, respectivement, une différence statistiquement significative (p<0,001).Le profil d'innocuité était favorable, aucun sujet n'ayant présenté d'inhibition du facteur F8, de malignité ou d'effets secondaires thrombotiques, et aucun événement indésirable grave (EIG) lié au traitement n'a été signalé.
3. Petits médicaments à base d'acide nucléique
(1) Vitravene
Société : Développée conjointement par Ionis Pharma (anciennement Isis Pharma) et Novartis.
Délai de mise sur le marché : Approuvé par la FDA et l'UE EMA en 1998 et 1999.
Indications : Pour le traitement de la rétinite à cytomégalovirus chez les patients séropositifs.
Remarques : Vitravene est un médicament oligonucléotidique antisens et le premier médicament oligonucléotidique approuvé pour la commercialisation dans le monde.Au début du marché, la demande du marché pour les médicaments anti-cytomégalovirus était très urgente ;puis du fait du développement de thérapies antirétrovirales hautement actives, le nombre de cas de cytomégalovirus a fortement chuté.En raison de la faible demande du marché, le médicament est sorti en 2002 et 2006 Retrait dans les pays de l'UE et aux États-Unis.
(2) Macugen
Entreprise : Co-développé par Pfizer et Eyetech.
Délai de mise sur le marché : Approuvé pour cotation aux États-Unis en 2004.
Indications : Pour le traitement de la dégénérescence maculaire néovasculaire liée à l'âge.
Remarques : Macugen est un médicament oligonucléotide modifié pégylé qui peut cibler et se lier au facteur de croissance endothélial vasculaire (isoforme VEGF165) et est administré par injection intravitréenne.
(3) Défini
Entreprise : Développé par Jazz.
Délai de mise sur le marché : Approuvé par l'Union européenne en 2013 et approuvé par la FDA en mars 2016.
Indications : Pour le traitement de la maladie occlusive des veinules hépatiques associée à un dysfonctionnement rénal ou pulmonaire après une greffe de cellules souches hématopoïétiques.
Remarques : Defitelio est un médicament oligonucléotidique, un mélange d'oligonucléotides ayant des propriétés de plasmine.Il a été retiré en 2009 pour des raisons commerciales.
(4) Kynamro
Entreprise : Co-développé par Ionis Pharma et Kastle.
Délai de mise sur le marché : Approuvé aux États-Unis en tant que médicament orphelin en 2013.
Indications : Pour le traitement adjuvant de l'hypercholestérolémie familiale homozygote.
Remarques : Kynamro est un médicament oligonucléotidique antisens, un oligonucléotide antisens ciblant l'ARNm de l'apo B-100 humaine.Kynamro est administré à raison de 200 mg par voie sous-cutanée une fois par semaine.
(5) Spinraza
Société : Développé par Ionis Pharmaceuticals.
Délai de mise sur le marché : Approuvé par la FDA en décembre 2016.
Indications : Pour le traitement de l'amyotrophie spinale (SMA).
Remarques : Spinraza (nusinersen) est un médicament oligonucléotidique antisens.Spinraza peut modifier l'épissage de l'ARN du gène SMN2 en se liant au site d'épissage de l'exon 7 de SMN2, augmentant ainsi la production de protéine SMN entièrement fonctionnelle.En août 2016, BIOGEN Corporation a exercé son option d'acquérir les droits mondiaux de Spinraza.Spinraza a commencé son premier essai clinique chez l'homme en 2011. En seulement 5 ans, il a été approuvé par la FDA en 2016, reflétant la pleine reconnaissance par la FDA de son efficacité.Le médicament a été approuvé pour la commercialisation en Chine en avril 2019. L'ensemble du cycle d'approbation de Spinraza en Chine est inférieur à 6 mois.Cela fait 2 ans et 2 mois que Spinraza a été approuvé pour la première fois aux États-Unis.Un tel nouveau médicament contre les maladies rares étrangères à succès est en La vitesse d'inscription en Chine est déjà très rapide.Cela est également dû à l'"Avis sur la publication de la première liste de nouveaux médicaments d'outre-mer nécessaires de toute urgence pour la recherche clinique" publié par le Centre d'évaluation des médicaments le 1er novembre 2018, qui a été inclus dans le premier lot de 40 nouveaux médicaments étrangers clés pour examen accéléré, et Spinraza s'y est classé.
(6) Exondys 51
Société : Développé par AVI BioPharma (rebaptisé plus tard Sarepta Therapeutics).
Délai de mise sur le marché : Approuvé par la FDA en septembre 2016.
Indications : Pour le traitement de la dystrophie musculaire de Duchenne (DMD) avec mutation du gène DMD dans le gène de saut de l'exon 51.
Remarques : Exondys 51 est un médicament oligonucléotidique antisens.L'oligonucléotide antisens peut se lier à la position de l'exon 51 du pré-ARNm du gène DMD, entraînant la formation d'ARNm mature.L'excision, corrigeant ainsi partiellement le cadre de lecture de l'ARNm, aide le patient à synthétiser certaines formes fonctionnelles de dystrophine plus courtes que la protéine normale, améliorant ainsi les symptômes du patient.
(7) Tegsédi
Société : Développé par Ionis Pharmaceuticals.
Délai de mise sur le marché : Approuvé par l'Union européenne pour la commercialisation en juillet 2018.
Indications : Pour le traitement de l'amylose héréditaire à transthyrétine (hATTR).
Remarques : Tegsedi est un médicament oligonucléotidique antisens ciblant l'ARNm de la transthyrétine.Il s'agit du premier médicament approuvé au monde pour le traitement de l'ATTRh.Le mode d'administration est l'injection sous-cutanée.Le médicament réduit la production de protéine ATTR en ciblant l'ARNm de la transthyrétine (ATTR) et présente un bon rapport bénéfice-risque dans le traitement de l'ATTR.Ni le stade de la maladie ni la présence d'une cardiomyopathie n'étaient pertinents.
(8) Sur Pattro
Entreprise : Co-développé par Alnylam et Sanofi.
Délai de mise sur le marché : Approuvé pour cotation aux États-Unis en 2018.
Indications : Pour le traitement de l'amylose héréditaire à transthyrétine (hATTR).
Remarques : Onpattro est un médicament siARN ciblant l'ARNm de la transthyrétine, qui réduit la production de protéine ATTR dans le foie et l'accumulation de dépôts amyloïdes dans les nerfs périphériques en ciblant l'ARNm de la transthyrétine (ATTR)., améliorant et soulageant ainsi les symptômes de la maladie.
(9) Givlaari
Entreprise : Développé par Alnylam Corporation.
Délai de mise sur le marché : Approuvé par la FDA en novembre 2019.
Indications : Pour le traitement de la porphyrie hépatique aiguë (PHA) chez l'adulte.
Remarques : Givlaari est un médicament à base d'ARNsi, le deuxième médicament à base d'ARNsi approuvé pour la commercialisation après Onpattro.Le médicament est administré par voie sous-cutanée et cible l'ARNm pour la dégradation de la protéine ALAS1.Un traitement mensuel par Givlaari peut réduire de manière significative et persistante le niveau d'ALAS1 dans le foie, réduisant ainsi les niveaux d'ALA et de PBG neurotoxiques à la normale, soulageant ainsi les symptômes de la maladie du patient.Les données ont montré que les patients traités par Givlaari présentaient une réduction de 74 % du nombre de poussées de la maladie par rapport au groupe placebo.
(10) Vyondys53
Entreprise : Développé par Sarepta Therapeutics.
Délai de mise sur le marché : Approuvé par la FDA en décembre 2019.
Indication : Pour le traitement des patients atteints de DMD présentant une mutation d'épissage de l'exon 53 du gène de la dystrophine.
Remarques : Vyondys 53 est un médicament oligonucléotidique antisens.Le médicament oligonucléotide cible le processus d'épissage du précurseur de l'ARNm de la dystrophine.Dans le processus indirect du précurseur de l'ARNm de la dystrophine, l'exon 53 externe a été partiellement épissé, c'est-à-dire non présent sur l'ARNm mature, et a été conçu pour produire une protéine dystrophine tronquée mais toujours fonctionnelle, améliorant ainsi la capacité d'exercice des patients.
(11) Waylivra
Société : Développé par Ionis Pharmaceuticals et sa filiale Akcea Therapeutics.
Délai de mise sur le marché : Approuvé par l'Agence européenne des médicaments (EMA) en mai 2019.
Indication : En tant que thérapie adjuvante à un régime contrôlé chez les patients adultes atteints du syndrome de chylomicronémie familiale (FCS).
Remarques : Waylivra est un médicament oligonucléotidique antisens, qui est le premier médicament approuvé pour le traitement du SCF dans le monde.
(12) Leqvio
Entreprise : Développé par Novartis.
Délai de mise sur le marché : Approuvé par l'UE en décembre 2020.
Indications : Pour le traitement de l'hypercholestérolémie primaire de l'adulte (hétérozygote familiale et non familiale) ou de la dyslipidémie mixte.
Remarques : Leqvio est un médicament siARN ciblant l'ARNm de PCSK9.Il s'agit de la première thérapie au monde par ARNsi hypocholestérolémiant (LDL-C).Le mode d'administration est l'injection sous-cutanée.Le médicament agit par interférence ARN pour abaisser les niveaux de protéine PCSK9, qui à son tour abaisse les niveaux de LDL-C.Les données cliniques montrent que Leqvio peut réduire le LDL-C d'environ 50 % chez les patients dont les taux de LDL-C ne peuvent pas être réduits aux niveaux cibles malgré les doses maximales tolérées de statines.
(13) Oxlumo
Société : Développé par Alnylam Pharmaceuticals.
Délai de mise sur le marché : Approuvé par l'UE en novembre 2020.
Indications : Pour le traitement de l'hyperoxalurie primaire de type 1 (PH1).
Remarques : Oxlumo est un médicament siARN ciblant l'ARNm de l'hydroxy acide oxydase 1 (HAO1), qui est administré par voie sous-cutanée.Le médicament a été développé à l'aide de la dernière technologie de conjugaison ESC-GalNAc chimique de stabilisation améliorée d'Alnylam, qui permet d'administrer des siARN par voie sous-cutanée avec une persistance et une efficacité accrues.Le médicament cible la dégradation ou l'inhibition de l'ARNm de l'hydroxyacide oxydase 1 (HAO1), réduit le niveau de glycolate oxydase dans le foie, puis consomme le substrat nécessaire à la production d'oxalate et réduit la production d'oxalate pour contrôler la progression de la maladie et améliorer les symptômes de la maladie chez les patients.
(14) Viltepso
Société : Développé par NS Pharma, une filiale de Nippon Shinyaku.
Délai de mise sur le marché : Approuvé par la FDA en août 2020.
Indications : Pour le traitement de la dystrophie musculaire de Duchenne (DMD) avec mutation du gène DMD dans le gène de saut de l'exon 53.
Remarques : Viltepso est un médicament oligonucléotide phosphorodiamide morpholino.Ce médicament oligonucléotidique peut se lier à la position de l'exon 53 du pré-ARNm du gène DMD, entraînant la formation d'ARNm mature.L'exon est partiellement éliminé, corrigeant ainsi partiellement le cadre de lecture de l'ARNm, aidant le patient à synthétiser certaines formes fonctionnelles de dystrophine plus courtes que la protéine normale, améliorant ainsi les symptômes du patient.
(15) Amvutra (vutrisiran)
Société : Développé par Alnylam Pharmaceuticals.
Délai de mise sur le marché : Approuvé par la FDA en juin 2022.
Indications : Pour le traitement de l'amylose héréditaire à transthyrétine chez l'adulte avec polyneuropathie (hATTR-PN).
Remarques : Amvuttra (Vutrisiran) est un médicament siARN ciblant l'ARNm de la transthyrétine (ATTR), qui est administré par injection sous-cutanée.Vutrisiran est conçu sur la base de la plate-forme d'administration conjuguée de la chimie de stabilisation améliorée (ESC)-GalNAc d'Alnylam avec une puissance et une stabilité métabolique accrues.L'approbation de la thérapie est basée sur les données de 9 mois de son étude clinique de phase III (HELIOS-A), avec des résultats globaux montrant que la thérapie améliorait les symptômes de hATTR-PN, avec plus de 50 % des patients inversant ou arrêtant la progression.
4. Autres médicaments de thérapie génique
(1) Rexine-G
Entreprise : Développé par Epeius Biotech.
Délai de mise sur le marché : Approuvé par la Philippine Food and Drug Administration (BFAD) en 2005.
Indications : Pour le traitement des cancers avancés résistants à la chimiothérapie.
Remarques : Rexin-G est une injection de nanoparticules chargées de gènes.Il introduit le gène mutant de la cycline G1 dans les cellules cibles par le biais d'un vecteur rétroviral pour tuer spécifiquement les tumeurs solides.Le mode d'administration est la perfusion intraveineuse.En tant que médicament ciblant les tumeurs qui recherche et détruit activement les cellules cancéreuses métastatiques, il a un certain effet sur les patients qui sont inefficaces contre d'autres médicaments anticancéreux, y compris les produits biologiques ciblés.
(2) Néovascularisation
Entreprise : Développé par l'Institut des cellules souches humaines.
Heure d'inscription : Approuvé pour l'inscription en Russie le 7 décembre 2011, puis inscrit en Ukraine en 2013.
Indications : Pour le traitement de la maladie artérielle périphérique, y compris l'ischémie grave des membres.
Remarques : Neovasculgen est une thérapie génique basée sur l'ADN plasmidique dans laquelle le gène du facteur de croissance endothélial vasculaire (VEGF) 165 est construit sur un squelette plasmidique et infusé aux patients.
(3) Collagène
Société : co-développée par l'Université d'Osaka et des sociétés de capital-risque.
Date d'inscription : Approuvé par le ministère japonais de la Santé, du Travail et du Bien-être pour l'inscription en août 2019.
Indications : Traitement de l'ischémie sévère des membres inférieurs.
Remarques : Collategene est une thérapie génique à base de plasmide, le premier médicament de thérapie génique natif japonais produit par AnGes.Le composant principal de ce médicament est un plasmide nu contenant la séquence génétique du facteur de croissance des hépatocytes humains (HGF).Si le médicament est injecté dans les muscles des membres inférieurs, le HGF exprimé favorisera la formation de nouveaux vaisseaux sanguins autour des vaisseaux sanguins occlus.Des essais cliniques ont confirmé son efficacité dans l'amélioration des ulcères.
FIN
Heure de publication : 10 novembre 2022



